



La pèrdua auditiva (HL) és la malaltia amb discapacitat sensorial més freqüent en humans.Als països desenvolupats, al voltant del 80% dels casos de sordesa prelingual en nens són causats per factors genètics.Els més comuns són els defectes d'un sol gen (com es mostra a la figura 1), s'han trobat 124 mutacions gèniques associades a la pèrdua auditiva no sindròmica en humans, la resta són causades per factors ambientals.Un implant coclear (un dispositiu electrònic col·locat a l'orella interna que proporciona estimulació elèctrica directament al nervi auditiu) és, amb diferència, l'opció més eficaç per tractar l'HL greu, mentre que un audiòfon (un dispositiu electrònic extern que converteix i amplifica les ones sonores) pot ajudar els pacients amb HL moderat.No obstant això, actualment no hi ha medicaments disponibles per tractar l'HL hereditari (GHL).En els últims anys, la teràpia gènica ha rebut una atenció creixent com a enfocament prometedor per tractar la disfunció de l'oïda interna.
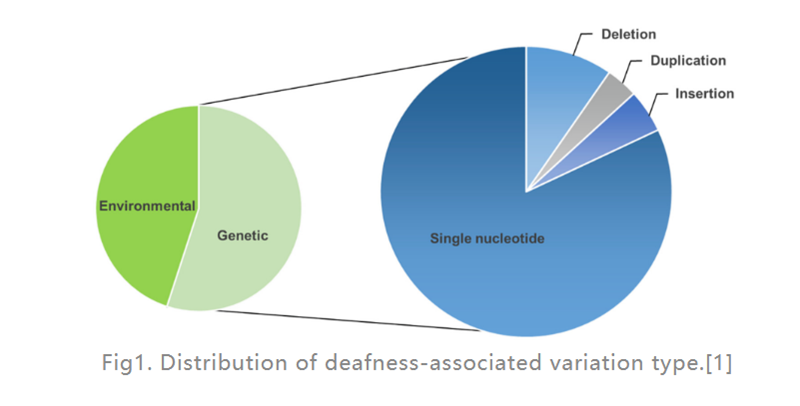
Fig1.Distribució del tipus de variació associada a la sordesa.[1]
Recentment, científics de l'Institut Salk i la Universitat de Sheffield van publicar un resultat d'investigació a Molecular Therapy - Methods & Clinical Development [2], que va mostrar àmplies perspectives d'aplicació per a la teràpia gènica in vivo de la sordesa hereditària.Uri Manor, professor ajudant d'investigació a l'Institut Salk i director del Centre Waitt per a Biofotònica Avançada, va dir que va néixer amb una pèrdua auditiva severa i va pensar que restaurar l'audició seria un regal meravellós.La seva investigació anterior va trobar que Eps8 és una proteïna reguladora de l'actina amb activitats d'unió i limitació de l'actina;a les cèl·lules ciliades coclears, el complex proteic format per Eps8 amb MYO15A, WHIRLIN, GPSM2 i GNAI3 existeix principalment en la majoria Les puntes dels estereocilis llargs, que juntament amb MYO15A localitzen BAIAP2L2 a les puntes dels estereocilis més curts, són necessàries per al manteniment dels paquets de cabell.Per tant, Eps8 pot regular la longitud dels estereocilis de les cèl·lules ciliades, que és essencial per a la funció auditiva normal;La supressió o la mutació d'Eps8 donarà lloc a estereocilis curts, que fan que no pugui convertir correctament el so en senyals elèctrics per a la percepció cerebral, que al seu torn condueix a la sordesa..Al mateix temps, el col·laborador Walter Marcotti, professor de la Universitat de Sheffield, va trobar que les cèl·lules ciliades no es poden desenvolupar amb normalitat en absència d'Eps8.En aquest estudi, Manor i Marcotti es van unir per investigar si afegir Eps8 a les cèl·lules estereociliars podria restaurar la seva funció i, al seu torn, millorar l'audició dels ratolins.L'equip d'investigació va utilitzar el vector del virus adenoassociat (AAV) Anc80L65 per lliurar la seqüència de codificació que conté EPS8 de tipus salvatge a la còclea de ratolins P1-P2 nounats Eps8-/- mitjançant injecció de membrana de finestra rodona;a les cèl·lules ciliades coclears del ratolí La funció dels estereocilis es va reparar abans que madurissin;i l'efecte de reparació es va caracteritzar per la tecnologia d'imatge i la mesura dels estereocilis.Els resultats van mostrar que Eps8 va augmentar la longitud dels estereocilis i va restaurar la funció de les cèl·lules ciliades a les cèl·lules de baixa freqüència.També van descobrir que, amb el temps, les cèl·lules semblaven perdre la seva capacitat de ser rescatades per aquesta teràpia gènica.La implicació és que aquest tractament pot haver de ser administrat a l'úter, ja que les cèl·lules ciliades Eps8-/- poden haver madurat o acumulat danys irreparables després del naixement dels ratolins."Eps8 és una proteïna amb moltes funcions diferents i encara hi ha molt per explorar", va dir Manor.La investigació futura inclourà investigar l'efecte de la teràpia gènica Eps8 en la restauració de l'audició en diferents etapes de desenvolupament i si és possible allargar les oportunitats de tractament.Casualment, el novembre de 2020, la professora KarenB Avraham de la Universitat de Tel Aviv a Israel va publicar els seus resultats a la revista EMBO Molecular Medicine [3], utilitzant una tecnologia innovadora de teràpia gènica per crear un virus sintètic adenoassociat AAV9-PHP inofensiu.B, El defecte del gen a les cèl·lules ciliades dels ratolins Syne4-/- es va reparar injectant un virus que portava la seqüència codificant de Syne4 a l'oïda interna dels ratolins, permetent-li entrar a les cèl·lules ciliades i alliberar el material genètic transportat, permetent-los madurar i funcionar amb normalitat (com a la figura 2).
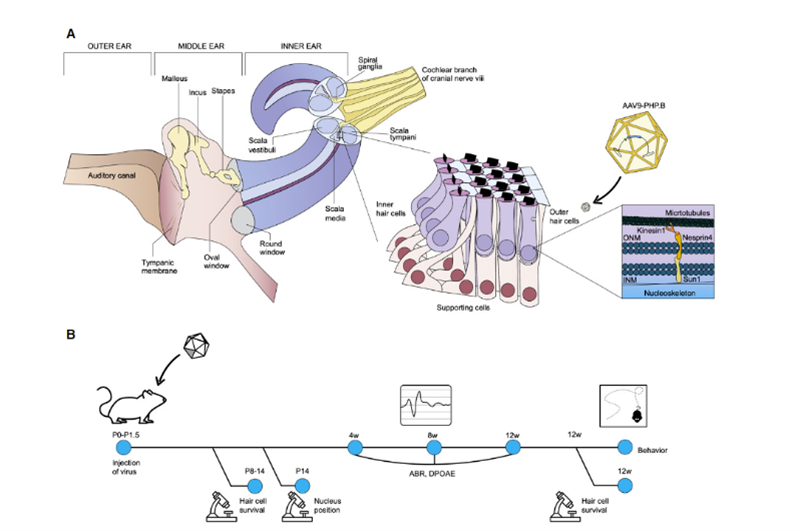
Fig2.Representació esquemàtica de l'anatomia de l'oïda interna, amb un focus en l'òrgan de Corti i la funció cel·lular de nesprin-4.
Es pot observar que l'ús de la teràpia gènica per aconseguir el propòsit de tractar malalties hereditàries a nivell genètic mitjançant la inserció, eliminació o correcció de qualsevol gen mutat per al tractament (és a dir, controlar els canvis genètics de la malaltia) té un efecte clínic elevat.perspectives d'aplicació.Els mètodes actuals de teràpia gènica per a la sordesa genèticament deficient es poden dividir en les següents categories:
substitució de gens
La substitució de gens és sens dubte la forma més "directa" de teràpia gènica, basada en identificar i substituir un gen defectuós per una còpia normal o salvatge del gen.Primer estudi reeixit de teràpia gènica de l'oïda interna per a la pèrdua auditiva causada per la supressió del gen del transportador de glutamat vesicular 3 (VGLUT3);El lliurament mediat per AAV1 de la sobreexpressió exògena de VGLUT3 a les cèl·lules ciliades de l'oïda interna (IHC) pot provocar una recuperació auditiva sostinguda, una recuperació parcial de la morfologia sinàptica de la cinta i respostes convulsives [4].Tanmateix, en els exemples que inclouen els dos reemplaçaments de gens lliurats per AAV descrits a la introducció anterior, és important tenir en compte que els models de ratolí utilitzats per a certs tipus de trastorns hereditaris de pèrdua auditiva de supressió de gens són temporalment diferents dels humans, i en els ratolins P1, l'oïda interna es troba en l'etapa madura de desenvolupament.En canvi, els humans neixen amb una orella interna madura.Aquesta diferència impedeix la possible aplicació dels resultats del ratolí al tractament dels trastorns hereditaris de la sordesa humana tret que la teràpia gènica s'administra a les orelles madures del ratolí.
Edició de gens: CRISPR/Cas9
En comparació amb el "reemplaçament de gens", el desenvolupament de la tecnologia d'edició de gens ha portat l'inici del tractament de les malalties genètiques des de l'arrel.És important destacar que el mètode d'edició de gens compensa les deficiències dels mètodes tradicionals de teràpia gènica amb sobreexpressió que no són adequats per a les malalties hereditàries dominants de sordesa i el problema que el mètode de sobreexpressió no dura gaire.Després que els investigadors xinesos van eliminar específicament l'al·lel mutant Myo6C442Y en ratolins Myo6WT/C442Y mitjançant el sistema d'edició del gen AAV-SaCas9-KKH-Myo6-g2, i dins dels 5 mesos posteriors a la eliminació, els ratolins es va restaurar la funció auditiva del model;al mateix temps, també es va observar que la taxa de supervivència de les cèl·lules ciliades a l'oïda interna es va millorar, la forma dels cilis es va regularitzar i es van corregir els indicadors electrofisiològics [5].Aquest és el primer estudi del món que utilitza la tecnologia CRISPR/Cas9 per al tractament de la sordesa hereditària causada per la mutació del gen Myo6, i és un important progrés de recerca de la tecnologia d'edició de gens per al tractament de la sordesa hereditària.La traducció clínica del tractament proporciona una base científica sòlida.
Mètodes de lliurament de teràpia gènica
Perquè la teràpia gènica tingui èxit, les molècules d'ADN nu no poden penetrar les cèl·lules amb eficàcia a causa de la seva hidrofilicitat i la seva càrrega negativa de grups fosfat, i per garantir la integritat de les molècules d'àcid nucleic complementades, s'ha de seleccionar un mètode segur i eficaç.L'ADN suplementat es lliura a la cèl·lula o teixit diana.L'AAV s'utilitza àmpliament com a vehicle de lliurament per al tractament de malalties a causa del seu alt efecte infecciós, baixa immunogenicitat i tropisme ampli per a diversos tipus de teixits.Actualment, un gran nombre de treballs de recerca han determinat el tropisme de diferents subtipus d'AAV en relació amb diferents tipus de cèl·lules a la còclea del ratolí.L'ús de característiques de lliurament d'AAV combinades amb promotors específics de la cèl·lula pot aconseguir l'expressió específica de la cèl·lula, que pot reduir els efectes fora de l'objectiu.A més, com a alternativa als vectors AAV tradicionals, es desenvolupen constantment nous vectors AAV sintètics i mostren una capacitat de transducció superior a l'oïda interna, dels quals AAV2/Anc80L65 és el més utilitzat.Els mètodes de lliurament no vírics es poden dividir en mètodes físics (microinjecció i electroporació) i mètodes químics (basats en lípids, polímers i nanopartícules d'or).Ambdós enfocaments s'han utilitzat en el tractament dels trastorns hereditaris de sordesa i han mostrat diferents avantatges i limitacions.A més del vehicle de lliurament de la teràpia gènica com a vehicle, es poden utilitzar diferents enfocaments per a l'administració de gens in vivo basats en diferents tipus de cèl·lules diana, vies d'administració i eficàcia terapèutica.La complexa estructura de l'oïda interna fa que sigui difícil arribar a les cèl·lules diana i la distribució dels agents d'edició del genoma és lenta.El laberint membranós es troba dins del laberint ossi de l'os temporal i inclou el conducte coclear, el conducte semicircular, l'utricle i el globus.El seu aïllament relatiu, la circulació limfàtica mínima i la separació de la sang per una barrera de laberint de sang limiten el lliurament sistèmic efectiu de terapèutica només als ratolins neonatals.Per obtenir títols virals adequats per a la teràpia gènica, és necessària la injecció local directa de vectors virals a l'oïda interna.Les vies d'injecció establertes inclouen [6]: (1) membrana de finestra rodona (RWM), (2) traqueotomia, (3) cocleostomia endolimfàtica o perilimfàtica, (4) membrana de finestra rodona més fenestració del tub (CF) (com a la figura 3).
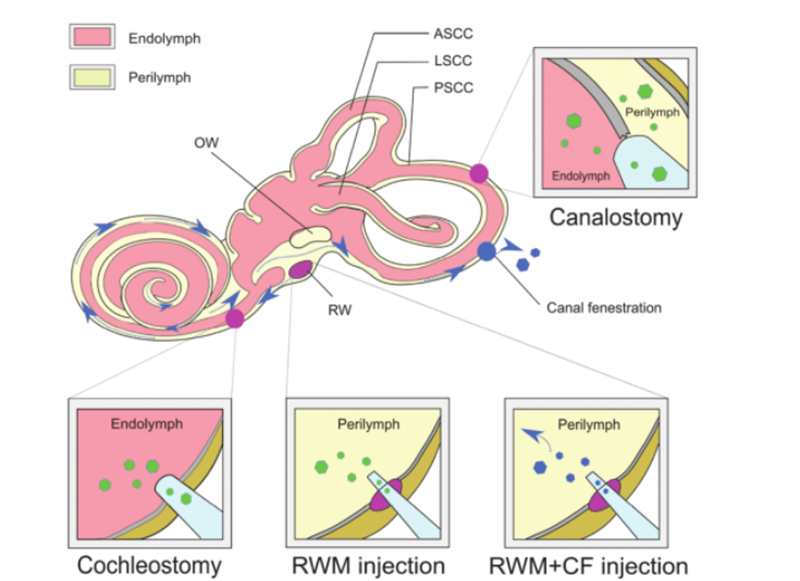
Fig3.Lliurament de la teràpia gènica a l'oïda interna.
Tot i que s'han fet molts avenços en la teràpia gènica, basats en objectius clínics de translació, cal treballar més abans que la teràpia gènica pugui esdevenir una opció de tractament de primera línia per als pacients amb malalties genètiques, especialment en el desenvolupament de vectors i mètodes de lliurament segurs i efectius.Però creiem que en un futur proper, aquest tipus de tractaments es convertiran en un element bàsic de la teràpia personalitzada i tindran un impacte molt positiu en la vida de les persones amb trastorns genètics i les seves famílies.
Foregene també ha llançat un kit de cribratge d'alt rendiment per a gens dirigits, que és ràpid i pot realitzar reaccions de transcripció inversa i qPCR sense extracció d'ARN.
Enllaços de productes
Kit Cell Direct RT-qPCR: Taqman/SYBR GREEN I
Per obtenir més informació sobre el producte, poseu-vos en contacte amb:
Hora de publicació: Set-02-2022



